Original Article |
|
Corresponding author: Nisha C. Patel ( patelnisha14785@gmail.com ) © 2022 Nisha C. Patel, Hitesh A. Patel.
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Patel NC, Patel HA (2022) A recent solidification approach for nanosuspension: formulation, optimisation and evaluation of canagliflozin immediate release pellets. Folia Medica 64(3): 488-500. https://doi.org/10.3897/folmed.64.e68866
|
Abstract
Introduction: Canagliflozin is a BCS class IV drug. Nanosuspension is known to enhance the saturation solubility and dissolution rate of poorly soluble drugs owing to the increased surface area of nanosized particles.
Aim: In the present study, we aimed to improve the dissolution characteristics of a poorly water-soluble drug canagliflozin by nanosuspension formulation and stability of this solubility enhancing system - nanosuspension can be improved by converting them into solidified forms as immediate release pellets.
Materials and methods: Canagliflozin nanosuspension was formulated using the media milling method. Poloxamer 407 was used to stabilise nanosuspension. Prepared nanosuspensions were subjected to the characterisation of particle size, polydispersity index (PDI), and drug content. Optimised nanosuspension (NS1) was solidified by converting into immediate release pellets: as improved stability, where canagliflozin nanosuspension was used as a binder. Pellets were prepared by +extrusion-spheronization technique using microcrystalline cellulose (MCC) as pelletizing aid and sodium starch glycolate as super disintegrant. Different important process parameters e.g. concentration of sodium starch glycolate (A), spheronization speed (B) and spheronization time (C) were investigated by 23 factorial design to accomplish desired disintegration time (R1) and drug release at 10 min (R2).
Results: The optimised nanosuspension had 120.5 nm particle size, 99.14% drug content and the optimised immediate release pellets (PF5) disintegrated within 23.29 second, and had 99.11% drug content. In vitro dissolution studies showed 89.59% drug release within 10 min in 0.75% w/v SLS. Scanning electron microscopy (SEM) confirmed uniform and spherically shaped pellets. Fourier transform infrared spectrometry (FTIR) and differential scanning calorimetry (DSC) analysis reveal no significant interaction between drug and excipients.
Conclusions: It can be concluded from the findings of this study that the formulation of nanosuspension and its use as a binder in the formulation of immediate release pellets should be investigated further in order to improve the dissolution rate and formulation stability.
Keywords
canagliflozin, extrusion, media milling, nanosized, spheronization
Introduction
Canagliflozin (CFZ) {(1S)-1, 5-anhydro-1-[3-[[5-(4-fluorophenyl)-2-thienyl]-methyl]-4-methylphenyl]-D-glucitol hemihydrate} is the first drug approved in this class of SGLT-2 inhibitors (also called gliflozin class of drugs).[
One of the major challenges of the pharmaceutical researchers is to develop an enabling formulation which can make poorly water-soluble drugs highly soluble to overcome the low bioavailability problem of the drugs. Many strategies have been used to improve the aqueous solubility of poorly soluble drugs such as complexation[
Nanotechnologies are one of the most prevalent strategies not only for overcoming the problem of poor solubility and thus bioavailability, but also for targeted drug delivery. Nanosuspensions are submicron dispersions of nanosized drug particles stabilised by surfactants, polymers, or a mixture of both.[
Pellets are a multiunit dosage form of fine powders or granules of bulk drugs and excipients. They range in size typically from about 0.5 mm to 1.5 mm of small, free flowing, spherical or semi-spherical solid units and are intended usually for oral administration. Extrusion-spheronization is a versatile method for obtaining pellets of high density, narrow particle size distribution, and high drug loading.[
In the present study, canagliflozin was used as a model drug and its nanosuspensions were prepared using the media milling method for the dissolution improvement. Optimised nanosuspension was used as a binder for the preparation of immediate release (IR) pellets, where MCC was used as a pelletizing agent and sodium starch glycolate as disintegrant and optimise the formula using the 23 factorial design by extrusion-spheronization technique. Physicochemical properties of nanosuspension including particle size and polydispersity index, and % drug content were evaluated. As the disintegration of the pellets leads to an increase in the surface area of the drug which is already in nanosize, stabilizing nanosuspension by converting it to immediate release pellets would improve the solubility, stability, and the dissolution rate of the drug
Aim
This study aims to improve the solubility of a drug by nanosuspension, and the transformation of nanosuspensions to the solid state ensures their long-term stability and increases patient compliance. Canagliflozin was used as a model drug and its nanosuspensions were prepared using the media milling method to improve the dissolution. An optimised nanosuspension was used as a binder for the preparation of immediate release (IR) pellets, where the MCC was used as a pelletizing agent and sodium starch glycolate (SSG) by extrusion-spheronization technique. The prepared pellets were optimised and evaluated for different characteristics and improved the dissolution compared to the pure drug and marketed product.
Materials and methods
Materials
Canagliflozin (gift sample from Zydus Cadila Pvt. Ltd, Ahmedabad). Poloxamer 407 (BASF Corporation, USA.) Zirconium oxide beads (0.5 mm) (gift sample from Synco Industries Limited, India.) Microcrystalline cellulose, corn starch, sodium starch glycolate and Kyron T 314 (gifts sample from Shiva Health Care, Mehsana). All chemicals we used were analytical and pharmaceutical grade. Distilled water was used throughout the study.
Methods
Preparation of canagliflozin nanosuspension
Nanosuspension of canagliflozin was prepared using wet media milling method.[
| Formulation code | Amount of drug mg | Amount of stabiliser mg | Amount of ZrO2 beads mg | Stirring time hour | Stirring speed rpm |
| NS1 | 100 | 100 | 8 | 16 | 1000 |
| NS2 | 100 | 100 | 8 | 16 | 600 |
| NS3 | 100 | 50 | 8 | 24 | 600 |
| NS4 | 100 | 50 | 4 | 24 | 1000 |
| NS5 | 100 | 50 | 4 | 16 | 600 |
Characterisation of canagliflozin nanosuspension
Particle size distribution and polydispersity index (PDI)
Particle size was determined by photon correlation spectroscopy using a Zetasizer 3000 (z-average, measuring range 20–1000 nm, Malvern Instruments, UK). All presented data are the means of the values for three independent samples obtained under identical conditions. Once the required intensity was reached, analysis was performed to get the mean particle size and polydispersity index(PDI).[
Drug content
An aliquot (1 ml) of the nanosuspension was diluted in methanol and diluted with 0.75% w/v of SLS, then filtered with a 0.2-μm filter. The total drug content was determined by UV spectrophotometer at λmax of the drug.[
Formula: 
Preparation of immediate release pellets
Pellets were prepared using the extrusion-spheronization technique.[
From the formulation and production result, it was observed that microcrystalline cellulose was used as pelletizing aids and sodium starch glycolate as super disintegrant, gave acceptable spherical pellets with good yield, low friability and satisfactory flow properties. Based on the observations, 30% of MCC, 10% of SSG and spheronization speed of 800 rpm with time 10 mm were selected as formulation and process parameters.
Formulation and optimisation of canagliflozin pellets by using 2 3 factorial design
In order to study the effect of processing and formulation variable on canagliflozin pellets, 23 factorial design was utilised by using Design Expert 12.0. This design consisted of 8 experimental trials (Table
R = β0+β1A+β2 B+β3C
| Formulation code | A: Conc. of SSG % | B: Spheronization speed rpm | C: Spheronization time min | MCC % w/w | Lactose % w/w | Drug mg |
| PF1 | 5 | 900 | 15 | 30 | 55 | 100 |
| PF2 | 5 | 600 | 15 | 30 | 55 | 100 |
| PF3 | 5 | 900 | 10 | 30 | 55 | 100 |
| PF4 | 5 | 600 | 10 | 30 | 55 | 100 |
| PF5 | 10 | 600 | 10 | 30 | 60 | 100 |
| PF6 | 10 | 600 | 15 | 30 | 60 | 100 |
| PF7 | 10 | 900 | 10 | 30 | 60 | 100 |
| PF8 | 10 | 900 | 15 | 30 | 60 | 100 |
Characterisation of drug loaded pellets
Determination of micromeritic properties [22]
Tap density and bulk density of the pellets were determined by tap density tester (Electro lab).
The angle of repose (θ) was measured by a fixed funnel method to know the flowability of the pellets.
Tan (θ) = h/r
where ‘h’ and ‘r’ are respectively the height and radius of the powder cone.
The Carr’s compressibility index (%) of the pellets was determined using following formula:
Carr’s Compressibility Index (%) = (Tapped density- Bulk density)/Tapped density
Hausner’s ratio of pellets was determined by ratio of tapped density to bulk density.
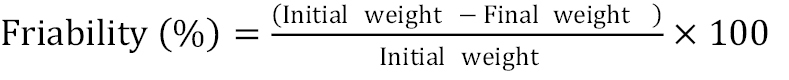
Friability is the measure of pellet strength. The friability of pellets was determined by using tablet friability tester (Electro lab) for a fixed period of time combined with glass beads of certain diameter in order to generate abrasion and to generate friability index. The pellets were then dedusted and reweighed.
Particle size and size distribution [21 ]
The size and size distribution of the pellets produced was determined by agitation for 10 min with a sieve shaker (Rotap LHC-41) fitted with a progression of standard sieves. From the weight retained on each sieve, particle size is determined from standard sieve aperture size as per Indian Pharmacopeia.
Disintegration time [22 ]
Disintegration time of pellets was determine as 100 mg pellets from each batch in distilled water by tablet disintegration tester (Electro lab) without disc at 37±0.5°C temperature with 30 dips speed. Disintegration test was carried out three times for the Immediate Release formulations and the results were expressed as mean ± standard deviation.
Drug content [22 ]
Accurately weighed 100 mg pellets were taken and converted in fine using mortar and pestle and the powder was dissolved in methanol using sonication and diluted with 0.75% w/v of SLS. The UV absorbance of the suitably diluted filtrate was measured at λmax 290 nm to determine the drug content. The concentration of drug was calculated using a calibration curve.
In vitro dissolution studies [13 ,19]
In vitro drug release studies of pellets were performed using the USP Apparatus I (basket) at a speed of 75 rpm in 900 ml of 0.75% w/v of SLS and the medium at 37.0±2°C. Canagliflozin is soluble in many organic solvents (methanol, dimethyl sulfoxide) and insoluble in aqueous media and freely soluble in 0.75% w/v of SLS.[
X-ray diffraction
[23 ]
X-ray diffractometer (Xpert MPD Philips, Holand) was used for qualitative powder x-ray diffraction of pure canagliflozin, physical mixture of excipients and final pellets (PF5). The instrument was operated at a voltage of 40 KV and a current of 30 mA, with copper as the tube anode material. The samples were run over a range of 2θ angles from 2° to 40°.
Fourier transform spectroscopy analysis
[24 ]
The infrared spectra of the samples were obtained using a compact Fourier transform infrared spectrometry (FT-IR) spectrometer ALPHA II (BRUKER, Germany). 1-2 mg of fine solid powder of sample was directly analysed over the region 400 to 4000 cm-1 in the instrument.
Differential scanning calorimetry [24 ]
The DSC thermogram was obtained by differential scanning calorimeter (DSC), on Shimadzu TA-60 model. The samples were hermetically sealed in an aluminum crucible before analysis. The system was purged with nitrogen gas at a flow rate of 40-50 ml/min. Heating was done between 50°C to 300°C at the rate of 10°C/min.
Scanning electron microscopy (SEM) [25 ]
The surface morphology and cross section of the optimised pellets was examined by scanning electron microscopy (Nova Nano SEM 450 FEI). The samples were scanned at a voltage of 20 kV with different magnification.
Results and discussion
Characterisation and optimisation of canagliflozin nanosuspension
Canagliflozin nanosuspensions were prepared using wet media milling method. The prepared nanosuspensions were clear and transparent due to reduced particle size. The effect of amount of stabiliser, amount of ZrO2 beads, spheronization time, and spheronization speed were studied on the attributes of nanosuspension. Particle size distribution is a very important parameter because reduced particle size helps in the improvement of solubility of pure drug, thereby increasing its dissolution rate (Table
The mean particle size of different batches of canagliflozin nanosuspension was found to be in the range of 120 nm to 300 nm. An optimised batch (NS1) of nanosuspension showed mean particle size of 120.5 nm (Fig.
The polydispersity index which is 0.217 in case of NS1 and for other formulation in the range of 0.131 to 0.307. This can be attributed to the good wetting property and dispersibility exhibited by the surfactant used in the above ratio. Therefore, the above concentration of the stabiliser was optimised. Impact of further higher ratios of surfactant on the particle size reduction and stability was not explored and can be further investigated. It seems reasonable to postulate that use of optimum concentration of suitable stabiliser and stirring rate results in fine nanosuspension with smaller and more uniform particle size.
Zeta potential of optimised canagliflozin loaded nanosuspension batch NS1 was found to be -23.0±4.75 mV which is sufficient for stability (Fig.
The drug content was determined by UV-visible spectrophotometer method. Formulation NS1 was found to contain the highest drug content amongst the other formulations which is 99.14%.
| Formulation code | Mean particle size (nm) * | Drug content (%)* | Polydispersity index (PDI) * |
| NS1 | 120.5±5.6 | 99.14±0.11 | 0.217±0.23 |
| NS2 | 242.6±7.4 | 92.57±0.25 | 0.254±0.58 |
| NS3 | 246.3±632 | 91.27±0.38 | 0.281±1.36 |
| NS4 | 179.6±4.4 | 95.42±0.21 | 0.131±0.84 |
| NS5 | 300±7.8 | 91.35±0.15 | 0.307±0.29 |

Evaluation of immediate release pellets
Results for the investigation of flow properties indicates good and excellent pellet properties for all the placebo batches. Addition of MCC as a pelletizing agent and sodium starch glycolate as disintegrant results in the improvement of flow. Results for flow properties and drug content of the prepared immediate release pellets factorial batches are presented in Table
Flow properties and drug content of the prepared immediate release pellets factorial batches
| Formulation code | Mean particle size µm | Angle of repose | Bulk density gm/ml | Tapped density gm/ml | Hausner’s ratio | Carr’s index | Friability % | Drug content % |
| PF1 | 900 | 29.24 | 0.76 | 0.79 | 1.03 | 3.79 | 0.58 | 91.24 |
| PF2 | 850 | 28.35 | 0.62 | 0.64 | 1.03 | 3.12 | 0.55 | 94.17 |
| PF3 | 1000 | 30.54 | 0.75 | 0.78 | 1.04 | 3.84 | 0.60 | 90.31 |
| PF4 | 700 | 27.63 | 0.61 | 0.65 | 1.06 | 6.55 | 0.51 | 95.36 |
| PF5 | 600 | 24.28 | 0.59 | 0.61 | 1.03 | 3.38 | 0.41 | 99.11 |
| PF6 | 800 | 27.21 | 0.60 | 0.65 | 1.08 | 8.33 | 0.48 | 96.84 |
| PF7 | 750 | 25.34 | 0.62 | 0.66 | 1.06 | 6.06 | 0.58 | 95.21 |
| PF8 | 850 | 25.87 | 0.64 | 0.69 | 1.07 | 7.25 | 0.57 | 94.75 |
Density and flow properties
Pellets prepared with MCC and SSG with all batches offered very good physical characteristics and flow properties. Density values for pellets shows that pellets prepared using formula as per batch PF5 produced dense pellets. Pellets prepared from all batch PF1 to PF8 have bulk density and tapped density in the range of 0.59 to 0.79. Due to the spherical shape, the pellets showed smaller angle of repose in the range of 24.28 to 30.54, which is indicative of excellent flowability. The pellets have also very good compressibility.
Friability
Friability is an essential parameter for any drug formulations especially for a potent drug. Pellets prepared within Batch PF5 demonstrated less friability compared to other batches. Pellets produced with different batches have higher friability in the range of 0.41–0.58 which is less than 1. Indicated low product loss because of material during spheronization process due to proper drying of pellets and high speed rotational movement that lead to few dust formation.
Drug content
From the results shown in Table
Particle size
The particle size of pellets was determined by sieve analysis. Set of standard sieves were arranged with progression of their sieve number. Based on fraction retained on each sieve average particle size was calculated that is described in Table
Disintegration time
Disintegration study revealed that the use of different concentration of disintegrants affects the process of disintegration. MCC used alone as pelletizing agent showed a slow disintegration process with a higher disintegrating time, also as the concentration of MCC reduced, the disintegration time also decreased. Addition of disintegrate further lowered the disintegration time (Table
Results of drug release for batches PF1 to PF8 are shown in Fig.
| Formulation code | A: Conc. of SSG % | B: spheronization speed rpm | C: spheronization time min | R1 disintegration time second * | R2 % drug release at 10 min % * |
| PF1 | 5 | 900 | 15 | 36.48±0.5 | 78.19±1.2 |
| PF2 | 5 | 600 | 15 | 30.54±0.24 | 80.28±0.2 |
| PF3 | 5 | 900 | 10 | 31.27±0.4 | 79.48±0.35 |
| PF4 | 5 | 600 | 10 | 28.52±0.87 | 84.26±0.58 |
| PF5 | 10 | 600 | 10 | 23.29±1.24 | 89.31±0.57 |
| PF6 | 10 | 600 | 15 | 25.34±0.68 | 84.23±1.34 |
| PF7 | 10 | 900 | 10 | 26.36±045 | 86.32±0.89 |
| PF8 | 10 | 900 | 15 | 32.46±0.24 | 80.68±0.54 |

Data analysis
The polynomial equations for full model relating to the response, disintegration time and drug release at 10 min, the transformed factor are shown in Table
Table
The contour and surface 3D plots (Fig.
It was observed that pellets prepared using nanosuspension (PF5) released more than 89% drug within 10 min as compared to the marketed tablet and pure drug which released only 24.63% and 18.65% drug, respectively within 10 min (Fig.
Solidification approach of nanosuspension by using it as a binder in immediate release pellets resulted in a marked increase in the drug release pattern, which can be due to the synergistic effect produced by the reduced particle size at nano level and delivery of the poorly soluble drug in the form of immediate release pellets. Therefore, it can be concluded that both approaches, i.e. use of nanosuspension as a binder and use MCC for immediate release pellets improved the dissolution rate of canagliflozin.
| Regression statistics | ||||
| R1 Disintegration time second | R2 Drug release at 10 min % | |||
| R² | 0.9451 | 0.9450 | ||
| Adjusted R² | 0.9039 | 0.9037 | ||
| Predicted R² | 0.7803 | 0.7800 | ||
| Mean standard error | 0.4688 | 0.4188 | ||
| Observation | 8 | 8 | ||
| Coefficient | P value | Coefficient | P value | |
| Intercept | 29.28 | 0.0056 | 82.84 | 0.0056 |
| A-conc. of SSG | -2.42 | 0.0067 | 2.29 | 0.0054 |
| B-spheronization speed | 2.36 | 0.0073 | -1.68 | 0.0161 |
| C-spheronization time | 1.92 | 0.0148 | -2.00 | 0.0088 |
| Equation: full model | DISINTEGRATION TIME = +29.28-2.42A+2.36B+1.92C | DRUG RELEASE AT 10 MIN = +82.84+2.29A-1.68B-2.00C | ||


X-ray diffraction
X-ray diffraction was used to analyse the potential changes in the inner structure of canagliflozin during the formulation. X-ray diffractogram of pure canagliflozin, physical mixture of excipients and final pellets (PF5) exhibited several peaks at different intensities between 2° to 40° (Fig.

Fourier transform spectroscopy analysis
The FTIR spectra of canagliflozin pellets (PF5) and nanosuspension (NS1) when compared with IR spectra of canagliflozin pure drug, showed no interaction of drug with excipient, indicating the compatibility of the drug with the excipient. Hence principle peaks of canagliflozin can be seen in the IR spectra of both formulation (Fig.

Differential scanning calorimetry (DSC)
The DSC thermogram of pure drug, physical mixture of all excipient and pellet batch (PB5) is shown in Fig.

Scanning electron microscopy
The morphology of optimised pellets PF5 analysed by scanning electron microscopy is shown in Fig.

Conclusions
On the basis of the results of the present study, we can conclude that the formulation of immediate release pellets using nanosuspension as a binder can be a unique and feasible option for stabilisation as well as solidification of nanosuspensions. Use of wet media milling method as a non-specific technique for size reduction can produce nanosuspension with significant improvement in dissolution rate. The study has demonstrated that it is possible to enhance the solubility of canagliflozin by preparing its nanosuspension using a media milling technique. Furthermore, the solubility of the drug can be improved by formulating immediate release pellets using MCC and SSG to improve the dissolution rate. Solidification of nanosuspension as pellets can be further explored for increasing the solubility of other poorly soluble drugs and stabilisation of nanosuspension.
Acknowledgements
The authors express their gratitude to Zydus Cadila Pvt. Ltd., Synco Industries Limited and Shiva Health Care for providing gift samples. The authors are thankful to the management of SSKM, Gujarat for providing the facilities to carry out the research work and Sophisticated lnstrumentation Gentre for Applied Research & Testing (SlCART) for providing facility of XRD and SEM.
Conflict of Interest
The authors declare no potential conflict of interest. The authors alone are responsible for the design of the study, the content and writing of the manuscript.
Author contributions
All authors have equal contribution to project work, data analysis and writing paper.
References
- 1. Compounds S: Canagliflozin Compound Summary for CID 24812758. PubChem open chemistry database: 2, 2016.
- 2. Haas B, Eckstein N, Pfeifer V, et al. Efficacy, safety and regulatory status of SGLT2 inhibitors: Focus on canagliflozin. Nutr Diabetes 2014; 4(11):1–8.
- 3. Lamos EM, Younk LM, Davis SN. Canagliflozin, an inhibitor of sodium-glucose cotransporter 2, for the treatment of type 2 diabetes mellitus. Expert Opin Drug Metab Toxicol 2013; 9(6):763–75.
- 4. Coelln-Hough J. Canagliflozin Advisory Committee Meeting. Janssen Pharm, 2013.
- 5. European Medicines Agency Assessment Report Vokanamet. Committee for Medicinal Products for Human Use. EMA/179391/2014; 2014.
- 6. Tahara A, Takasu T, Yokono M, et al. Characterization and comparison of sodium-glucose cotransporter 2 inhibitors in pharmacokinetics, pharmacodynamics, and pharmacologic effects. J Pharmacol Sci 2016; 130(3):159–69.
- 7. Dietrich E, Powell J, Taylor JR. Canagliflozin: a novel treatment option for type 2 diabetes. Drug Des Devel Ther 2013; 7:1399–08.
- 8. European Medicines Agency, Assessment Report Canagliflozin. Committee for Medicinal Products for Human Use, EMA/374133/2013: 2013.
- 9. Liversidge GG, Cundy KC. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs. Int J Pharm 1995; 125:91–97.
- 10. Javiana L, Teobaldo A, Jacqueline S, et al. Preliminary pharmacokinetic study of different preparations of acyclovir with b-cyclodextrin. J Pharm Sci 2002; 91:2593–8.
- 11. Norbert R, Muller BW. Dissolution rate enhancement by in situ micronization of poorly water-soluble drugs. Pharm Res 2002; 19:1894–900.
- 12. Hajare AA, Jadhav PR. Improvement of solubility and dissolution rate of indomethacin by solid dispersion in polyvinyl pyrrolidone K30 and poloxomer 188. Asian J Pharm Technol 2012; 2:116–22.
- 13. Ubgade S, Kilor V, Bahekar V, et al. Formulation development of immediate release pellets of tadalafil: solidification approach for nanosuspension. In J Appl Pharm 2019; 11(4):124–131.
- 14. Asare Addo K, Supuk E, Al Hamidi H, et al. Triboelectrification and dissolution property enhancements of solid dispersions. Int J Pharm 2015; 485:306–16.
- 15. Hafner A, Lovrić J, Lakoš GP, et al. Nanotherapeutics in the EU: an overview on current state and future directions. Int J Nanomedicine 2014; 9:1005–23.
- 16. Erkoboni KA. In: Ghebre-Sellassie I, Martin C, editors. Pharmaceutical Extrusion Technology. Extrusion/spheronization. New York: Marcel Dekker Inc; 2003:277–322.
- 17. Fielden KE, Newton JM, Rowe RC. The influence of lactose particle size on spheronization of extrudate processed by a ram extruder. Int J Pharm 1992; 81:205–24.
- 18. Patel H, Patel H, Gohel M, et al. Dissolution rate improvement of telmisartan through modified MCC pellets using 32 full factorial design. Saudi Pharm J 2016; 24(5):579–87.
- 19. Patel NC, Patel HA. Application of Plackett-Burman screening design in optimization of process parameters for formulation of Canagliflozin nanosuspension. Int J Pharm Sci Nanotech 2020; 13(6):5208–16.
- 20. Shid RL, Dhole SN, Kulkarni N, et al. Formulation and evaluation of nanosuspension formulation for drug delivery of simvastatin. Int J Pharm Sci Nanotech 2014; 7(4):2650–65.
- 21. Law MF, Deasy PB. Use of hydrophilic polymers with microcrystalline cellulose to improve extrusion-spheronization. Eur J Pharm Biopharm 1998; 45(1):57–65.
- 22. Shelar S, Shirolkar S, Kale N. Formulation optimization of promethazine theoclate immediate release pellets by using extrusion-spheronization technique. Int J Appl Pharm 2018; 10(1):30–35.
- 23. Ruchi A, Nirav P, Mihir R. Novel amorphous solid dispersions of canagliflozin hemihydrate in EUDRAGIT® E PO. Int J Pharm Sci Res 2019; 10(6):2923–33.
- 24. Bhatta R, Rudragangaiah S, Babu S, et al. Formulation, optimization and evaluation of in-situ gelling liquid oral formulation of a novel antidiabetic drug: canagliflozin. Indian J Pharm Educ Res 2019; 53(2):121–8.
- 25. Arora U, Thakkar V, Baldaniya L, et al. Fabrication and evaluation of fast disintegrating pellets of cilostazol. Drug Dev Ind Pharm 2020; 46(12):1927–46.